This toolkit is open source. Any third-party models, product names, or trademarks referenced are the property of their respective owners, and Proto is not affiliated with them.
martinpacesa/BindCraft
One-shot design of functional protein binders with BindCraft
Open Notebook
Open notebook
Run locally with proto-tools



License: BindCraft’s own code is licensed under MIT, but it runs as a pipeline that depends on bundled components and model weights under separate license terms, including non-commercial or restricted-use terms. The bundled model weights are licensed under CC-BY-4.0. As a whole the pipeline has restrictions around commercial use and may require explicit attribution when utilized.Bundled dependencies, each under its own license:
- PyRosetta: Custom (PyRosetta Software License)
Background
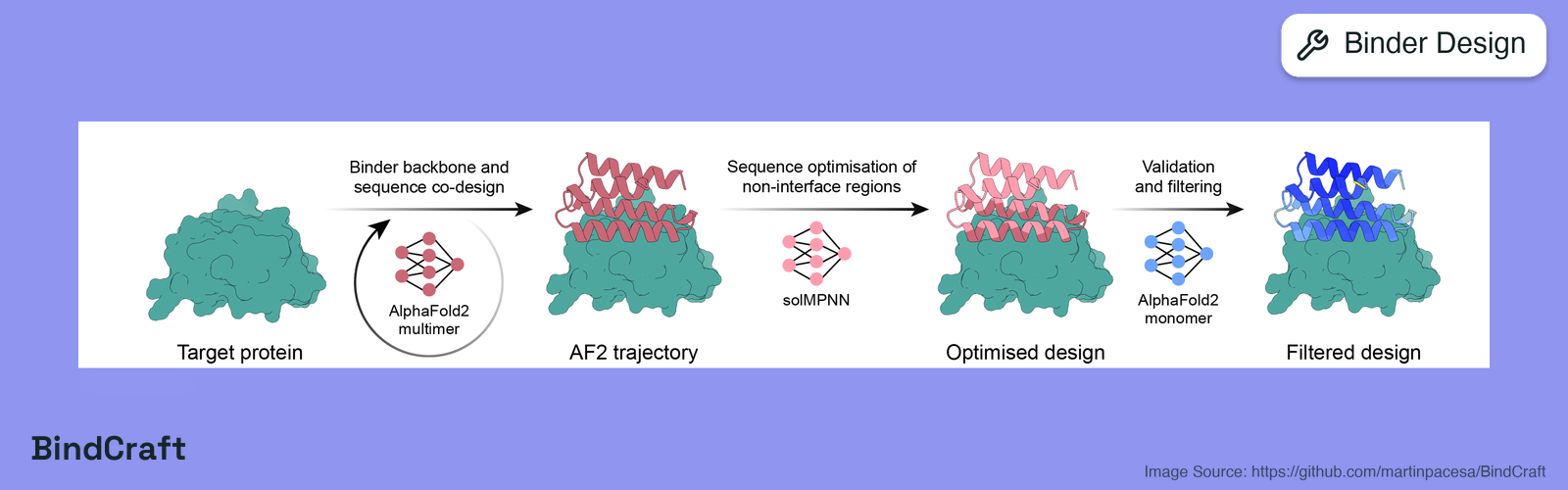
BindCraft (Pacesa et al., 2025) addresses the problem of generating protein binders against a target without the need for high-throughput experimental screening or curated structural templates. The published pipeline reports experimental success rates of 10 to 100 percent across diverse and challenging targets including cell-surface receptors, common allergens, de novo designed proteins, and multi-domain nucleases such as CRISPR-Cas9, and produces binders with nanomolar affinity. The authors demonstrate functional and therapeutic applications including reduction of IgE binding to birch allergen in patient-derived samples, modulation of Cas9 gene editing activity, and reduction of cytotoxicity from a foodborne bacterial enterotoxin. The pipeline chains four stages per design trajectory. First, an AlphaFold2 hallucination step initialises a binder of randomly sampled length adjacent to the frozen target and optimises the binder logits by gradient descent against a weighted sum of structural losses that includes per-residue pLDDT, intra-binder and inter-chain PAE, intra-binder and interface contact counts, interface pTM, a helicity bias, and a radius-of-gyration term. Second, the hallucinated backbone is handed to ProteinMPNN (Dauparas et al., 2022) which samples a set of foldable sequences while optionally holding interface residues fixed. Third, each ProteinMPNN-refined complex is re-predicted from scratch with AlphaFold2 multimer (Jumper et al., 2021) as an independent validation of the design. Fourth, the validated complex is relaxed with PyRosetta and scored against an extensive set of interface metrics including binding-energy difference, shape complementarity, buried surface area, hydrogen bond counts, packing statistic, secondary-structure composition, and hotspot RMSD. A trajectory is accepted only when every metric clears the corresponding upstream filter threshold.Learning Resources
- martinpacesa/BindCraft (Correia Lab, EPFL). Official BindCraft repository, command-line interface, and reference filter configurations.
- BindCraft tutorial notebook (Correia Lab). Walkthrough of the design pipeline with pre-set example targets and parameter explanations.
Tools
BindCraft Binder Design (bindcraft-design)
Designs one or more de novo protein binders against a user-supplied target. The tool takes a target structure together with the target chain identifiers, an optional hotspot residue list, and a binder length range, and runs the BindCraft pipeline until either the requested number of accepted designs has been produced or the configured trajectory limit has been reached. The output carries each accepted binder as an amino-acid sequence, a relaxed target-binder complex Structure with per-residue pLDDT in the B-factor column, and the per-design BindCraft metrics used by the filter check.API Reference
Source
Input: BindCraftInput
Input: BindCraftInput
Structure
required
Target structure. Accepts a file path, raw PDB/CIF content string,
Structure object, or a dict in the shape produced by Structure.model_dump(mode='json').string
default:"A"
Chain ID(s) of the frozen target (comma-separated for multi-chain). Maps to BindCraft’s
chains.string
Comma-separated 1-indexed residue positions on the target that the binder must contact. Supports ranges (e.g.
"1-10,56,78"). None or empty = unrestricted.array
default:"[65, 150]"
(min, max) binder length range. Maps to BindCraft’s lengths.string
default:"binder"
Project identifier — used as a prefix in output filenames.
integer
default:"100"
Target accepted-design count. The pipeline stops after reaching this count or after
max_trajectories attempts (whichever comes first). Source
Config: BindCraftConfig
Config: BindCraftConfig
enum
default:"4stage"
Hallucination algorithm. Drives which iteration-count fields below are actually consumed (see each field’s
depends_on). UpstreamAvailable options: 2stage, 3stage, 4stage, greedy, mcmcboolean
default:"True"
Use AF2 multimer parameters during hallucination. Every upstream preset uses multimer.
string
default:"C"
Amino acids to ban during design (no separator). Upstream default:
"C".boolean
default:"False"
Reject any design containing
omit_AAs.integer
default:"75"
Soft-stage iterations. Used by 2stage/3stage/4stage.
integer
default:"45"
Temporary-stage iterations. Used by 3stage/4stage.
integer
default:"5"
Hard-stage iterations. Used by 3stage/4stage.
integer
default:"15"
Greedy/MCMC iterations. Used by 2stage/4stage/greedy/mcmc.
number
default:"1.0"
Greedy/MCMC mutation rate as % of binder length.
number
default:"0.1"
pLDDT loss weight.
number
default:"0.4"
Intra-chain PAE loss weight.
number
default:"0.1"
Inter-chain (interface) PAE loss weight.
number
default:"1.0"
Intra-chain contact loss weight.
number
default:"1.0"
Inter-chain (interface) contact loss weight.
number
default:"-0.3"
Helicity bias weight (negative discourages helices).
number
default:"0.05"
Interface pTM loss weight (only used when
use_i_ptm_loss=True).number
default:"0.3"
Radius-of-gyration loss weight (only used when
use_rg_loss=True).number
default:"0.1"
N-/C-termini distance loss weight (only used when
use_termini_distance_loss=True).boolean
default:"False"
Randomize the sign of
weights_helicity per trajectory.boolean
default:"True"
Enable interface pTM loss.
boolean
default:"True"
Enable radius-of-gyration loss.
boolean
default:"False"
Enable termini-distance loss.
number
default:"14.0"
Intra-chain contact distance cutoff (Å).
number
default:"20.0"
Inter-chain contact distance cutoff (Å).
integer
default:"2"
Number of intra-chain contacts per residue.
integer
default:"2"
Number of inter-chain contacts per residue.
boolean
default:"False"
Mask target template sequence during hallucination.
boolean
default:"False"
Mask target template sequence during validation.
boolean
default:"False"
Mask target template side chains during hallucination.
boolean
default:"False"
Mask target template side chains during validation.
boolean
default:"False"
Use the trajectory structure as AF2’s initial guess.
boolean
default:"False"
Use AF2’s “Big Bang” recycle initialisation.
boolean
default:"True"
Run ProteinMPNN sequence refinement after each accepted trajectory. When False, the
mpnn_* / num_seqs / max_mpnn_sequences / sampling_temp / backbone_noise / model_path fields are inert.boolean
default:"True"
Fix interface residues during MPNN redesign.
integer
default:"20"
Number of MPNN sequences to sample per trajectory.
integer
default:"2"
Max MPNN sequences to validate per trajectory.
number
default:"0.1"
MPNN sampling temperature (lower = more deterministic).
number
default:"0.0"
MPNN backbone noise.
enum
default:"v_48_020"
MPNN model checkpoint name.Available options:
v_48_002, v_48_010, v_48_020, v_48_030enum
default:"soluble"
MPNN weight set.Available options:
original, solubleinteger
default:"1"
AF2 recycles during hallucination.
integer
default:"3"
AF2 recycles during validation.
boolean
default:"True"
4stage-only — increase recycles + iterations mid-trajectory when the soft-stage output is beta-heavy.
integer
default:"0"
Extra soft iterations for beta-heavy designs.
integer
default:"0"
Extra temporary iterations for beta-heavy designs.
integer
default:"3"
Recycles during hallucination for beta-heavy designs.
integer
default:"3"
Recycles during validation for beta-heavy designs.
integer | boolean
default:"False"
Max hallucination trajectories before stopping.
False (upstream default) = unlimited; positive int = cap.boolean
default:"True"
Enable rolling acceptance-rate monitoring (stops the run if it stalls).
number
default:"0.01"
Minimum design acceptance rate to keep running.
integer
default:"600"
Trajectory count before acceptance-rate monitoring starts.
Dict[string, any]
Per-metric threshold overrides merged on top of the upstream default filters at dispatch time. Keys are upstream metric names (e.g.
"Average_pLDDT"); values are upstream filter dicts (e.g. {"threshold": 0.85, "higher": True}).integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cuda"
Device to run the tool on.
integer
Maximum execution time in seconds.
None (default) waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.Applications
This tool is appropriate for de novo binder generation against a structurally characterised target where no curated antibody scaffold or pre-existing binder is available. Representative applications include designing miniprotein binders against cell-surface receptors, generating binders that occlude a specific epitope or active site through hotspot targeting, producing structurally diverse binder candidates for downstream therapeutic engineering, and benchmarking AlphaFold2-hallucination as a binder discovery method against alternative approaches.Usage Tips
- Provide a hotspot residue list when targeting a defined epitope. Set
target_hotspot_residuesto a comma-separated list of residue positions on the target structure, with ranges supported (for example"1-10,56,78"). Residue numbering is 1-indexed to match standard biological residue numbering conventions. Without hotspots the binder may land anywhere on the target surface. With hotspots, BindCraft biases the hallucination loss to bring the binder into contact with the specified residues. Choose functional residues such as active sites, paratope contacts, or catalytic loops rather than arbitrary surface positions. binder_lengthsdefaults to(65, 150)residues, matching the upstream default. Binders below approximately 50 residues are effectively peptides and the AlphaFold2 multimer signal weakens. Binders above approximately 200 residues introduce significant GPU memory and per-trajectory runtime costs. Choose a tighter range to focus a campaign on a specific binder size class.weights_helicitycontrols the helix bias during hallucination. The default of-0.3is a mild anti-helix bias chosen by the upstream authors because AlphaFold2 tends to over-produce alpha-helical bundles. Set a positive value to encourage helices for helix-friendly targets, or setrandom_helicity=Trueto randomise the sign per trajectory and increase secondary-structure diversity across the campaign.optimise_beta=True(the default) adds extra hallucination iterations and AlphaFold2 recycles when a trajectory looks beta-heavy. Keep this enabled for any target that may favour beta-strand interfaces, such as immunoglobulin folds. The behaviour is gated on detected sheet content during the trajectory.filter_overrideslets you relax or tighten individual filter thresholds. Pass a dict keyed by upstream metric name (such as"Average_i_pTM") and valued as a filter dict ({"threshold": 0.45, "higher": True}). Only the listed metrics are overridden; every other filter keeps its upstream default. Lower the interface pTM or shape complementarity threshold first if zero designs are accepted on a hard target.- Production runs use
number_of_final_designs=100andmax_trajectories=False. This is the upstream default and produces enough accepted designs for downstream triage and experimental ordering. For a smoke test, set both to1together with reduced iteration counts (for examplesoft_iterations=10,temporary_iterations=5,hard_iterations=2,greedy_iterations=2) to verify the install and produce a single sample. enable_rejection_check=True(the default) aborts a run early if the rolling acceptance rate falls belowacceptance_rate=0.01afterstart_monitoring=600trajectories. Disable this gate when working on stubborn targets where you are willing to grind through many failed trajectories before the first acceptance.- The output is iterable. Iterating directly over the returned
BindCraftOutputyields each acceptedBindCraftDesignin turn, andlen(result)returns the number of accepted designs. - Complementary tools cover adjacent design tasks. Reach for
proteinmpnn-samplewhen an existing target-bound binder backbone only needs sequence redesign,alphafold2-gradient(with the Germinal backend) or a dedicated antibody-design pipeline for CDR-only redesign on a fixed antibody framework,rfdiffusion3-designwhen only a backbone is required without an accompanying sequence, and chemistry-aware ligand generation, docking, and scoring tools when the target is a small-molecule ligand rather than a protein binder.
Toolkit Notes
These apply to every BindCraft tool in this toolkit (bindcraft-design).
- The pipeline runs on a single GPU per trajectory and benefits from 32 to 80 GB of GPU memory. AlphaFold2 multimer dominates the memory footprint and scales with the combined target plus binder length. For targets larger than approximately 2000 residues, trim the target to its binder-accessible domain before running. To parallelise across multiple GPUs, run multiple instances of
bindcraft-designconcurrently through aToolPool. - The first run downloads approximately 5.5 GB of AlphaFold2 weights together with the ColabDesign, ProteinMPNN, and BindCraft repositories. Subsequent runs reuse the cached weights, which are shared with the proto-tools
alphafold2toolkit.
Infrastructure Guides
The following guides cover how to run tools efficiently and at scale.Tool Persistence
Keep a tool’s model warm across calls instead of reloading it every invocation.
Device Management
How GPUs are allocated to tools and how to target specific devices.
Parallel Execution
Fan a batch of inputs out across multiple GPUs.