 IPD
IPDLicense: RoseTTAFold3 (RF3) is open source and free for academic and commercial use under a BSD-3-Clause license and may require explicit attribution when utilized. Please refer to the license for full terms.
Proto is not affiliated with Institute for Protein Design. This toolkit is open source and builds on the implementation produced by this organization. Product names, logos, and trademarks are the property of their respective owners.
RosettaCommons/foundry
Accelerating Biomolecular Modeling with AtomWorks and RF3
Open Notebook
Open notebook



Background
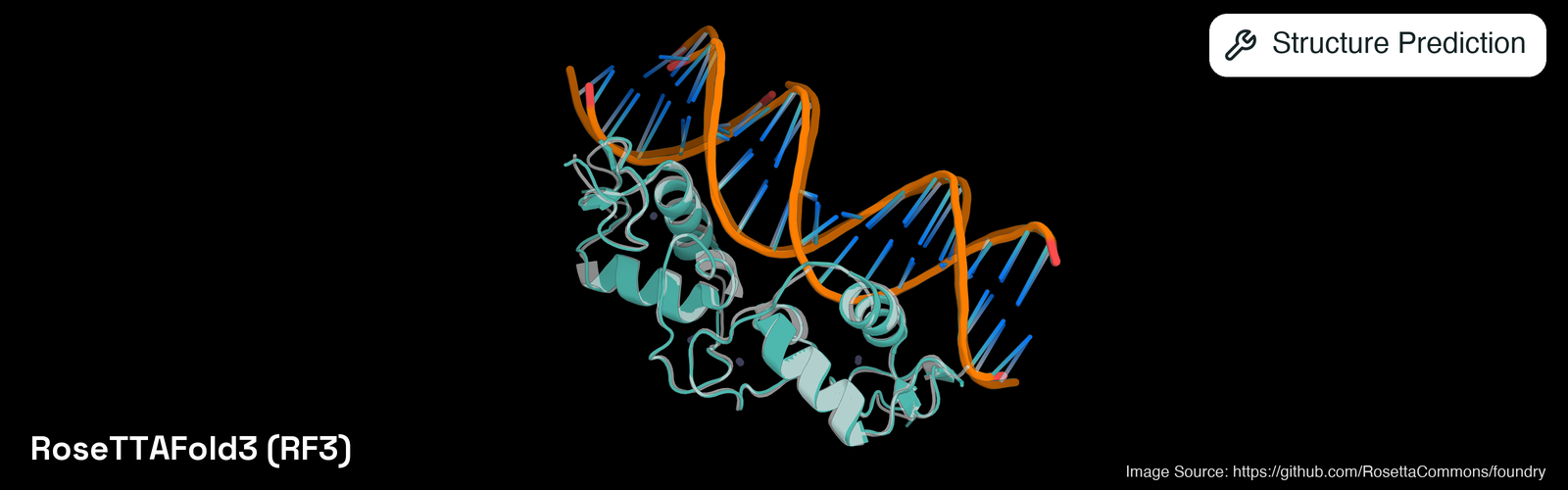
RoseTTAFold3 (Corley et al., 2025) predicts the joint 3D structure of a biomolecular assembly from the sequences and chemical components it contains. It is the latest entry in the RoseTTAFold lineage from the Baker and DiMaio labs at the Institute for Protein Design, and like AlphaFold3 and Boltz-2 it folds proteins, nucleic acids, and small-molecule ligands within a single model. The preprint reports that an improved treatment of chirality narrows the performance gap between RF3 and the closed-source AlphaFold3 on biomolecular benchmarks. Architecturally RF3 builds on the new AtomWorks data framework introduced alongside it in the preprint, and uses an AlphaFold3 style trunk together with a diffusion module that samples several candidate structures per complex from random noise. The best sample is selected by a composite ranking score that combines interface pTM, overall pTM, and a clash penalty. Alongside the predicted coordinates, RF3 reports per-residue and overall pLDDT, per-chain confidence, predicted aligned error (PAE) and predicted distance error (PDE), chain-pair PAE and PDE matrices for multi-chain inputs, and a boolean flag for steric clashes. The reference implementation is open-sourced as part of the RosettaCommons/foundry monorepo under the BSD-3-Clause license, with model weights served openly from the IPD file server. It was developed at the Institute for Protein Design (UW).Tools
RoseTTAFold3 Prediction (rf3-prediction)
Predicts the 3D structure of a biomolecular complex. Each input complex can combine protein, DNA, RNA, and ligand chains. The assembly is folded by RF3 and returned as a predicted Structure per complex with confidence metrics, including average pLDDT, pTM, interface pTM for multi-chain inputs, per-chain pTM, an overall and chain-pair PAE and PDE in angstroms, a composite ranking score, and a steric-clash flag.API Reference
Source
Input: RF3Input
Input: RF3Input
List[Complex]
required
List of complexes to predict structures for. Inherited from
StructurePredictionInput. Each complex can contain multiple chains of proteins, DNA, RNA, and/or ligands.array
Pre-computed MSAs, one entry per complex. Each entry is a
ComplexMSAs (per-chain MSAs keyed by chain index); paired=True marks rows taxonomy-aligned across chains. Populated by preprocess() or supplied directly. Default: None. Source
Config: RF3Config
Config: RF3Config
integer
default:"10"
Iterative refinement passes through the network. Higher = more accurate but slower. Default 10 (upstream default).
integer
default:"5"
Independent diffusion samples drawn per complex; the best by
ranking_score is returned. Default 5.integer
default:"50"
Denoising steps in the diffusion process. Default 50.
List[string]
Chain IDs (e.g.
["A"]) to mark as cyclic. Default [].integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr). Inherited from
BaseConfig. Default 0.string
default:"cuda"
"cuda" or "cpu". Inherited. Default "cuda".integer
default:"1800"
Maximum execution time in seconds. RF3 is heavier than Boltz2; the default is set accordingly. Default 1800.
integer
Inherited. Default
None.boolean
default:"False"
Inherited. Must remain False for RF3 (no per-token PAE matrix is emitted).
boolean
default:"True"
Generate MSAs for protein chains via MMseqs2 homology search. Supplied MSAs are always used and override
use_msa=False. Inherited from MSAStructurePredictionConfig. Default True.Mmseqs2HomologySearchConfig
Inherited. Default
None.boolean
default:"True"
Use taxonomy-paired MSA generation for heterocomplex protein chains. Inherited. Default
True. Source
Metrics (one set per
Output: RF3Output
Output: RF3Output
List[Structure]
required
Predicted structures, each carrying a :class:
RF3Metrics instance on .metrics.structures item)Applications
This tool predicts the structure of multi-component assemblies such as protein-DNA and protein-RNA complexes or protein-ligand binding poses. Within this toolkit it is also the model whose architecture has explicit chirality representations built in, which is relevant when modelling chiral small molecules, D-amino-acid residues, or peptides where stereochemistry matters. For multi-chain inputs the reported chain-pair PAE and PDE matrices together with interface pTM estimate how confidently the components are placed relative to each other, useful for ranking or filtering predicted interfaces.Usage Tips
use_msadefaults toTrue. A ColabFold search generates an MSA for each protein chain. Set itFalsefor single-sequence prediction, or attach precomputed MSAs to the input.- Diffusion samples are ranked by
ranking_score.diffusion_batch_size(default5) independent samples are drawn per complex. The best byranking_score = 0.8*iptm + 0.2*ptm - 100*has_clashis returned, withnum_steps(default50) controlling the denoising step count. n_recycles(default10) trades accuracy for time. More recycling iterations refine the prediction at higher runtime. Leave the upstream default of10unless you have a specific reason to lower it.- Cyclic chains. Mark chains as cyclic (head-to-tail) with
cyclic_chains=["A", ...]. - No template or conformer conditioning. RF3 can condition on input coordinates (templates, holo ligand conformers), but this wrapper accepts only sequences, SMILES, and CCD codes — no coordinate input — so those upstream options are not exposed.
- No per-token PAE matrix. Unlike Boltz-2 and AlphaFold3, RF3 emits only chain-pair PAE aggregates (
avg_pae,chain_pair_pae,chain_pair_pae_min) and a separatepde(predicted distance error). The inheritedinclude_pae_matrixtoggle is rejected byRF3Config. - Multi-modal inputs. Protein, DNA, RNA, and ligand entities are supported.
Toolkit Notes
These apply to every RF3 tool in this toolkit (rf3-prediction).
- Requires a GPU. RF3 runs through a PyTorch backend and needs an NVIDIA GPU. CPU execution is not practical.
- Open weights. The RF3 checkpoint is downloaded automatically from the IPD file server during environment setup and lands in the proto-tools weights cache. No request form or token is required.
- Predictions are stochastic. Structures come from a diffusion process, so repeated runs vary unless sampling is seeded. The wrapper advances the seed per complex within a batch so duplicate inputs in one call still diversify.
Infrastructure Guides
The following guides cover how to run tools efficiently and at scale.Tool Persistence
Keep a tool’s model warm across calls instead of reloading it every invocation.
Device Management
How GPUs are allocated to tools and how to target specific devices.
Parallel Execution
Fan a batch of inputs out across multiple GPUs.