License: PyHMMER is open source and free for academic and commercial use under an MIT license. Please refer to the license for full terms.
This toolkit is open source. Any third-party models, product names, or trademarks referenced are the property of their respective owners, and Proto is not affiliated with them.
pyHMMER: a Python library binding to HMMER for efficient sequence analysis
Open Notebook
Open notebook



Background
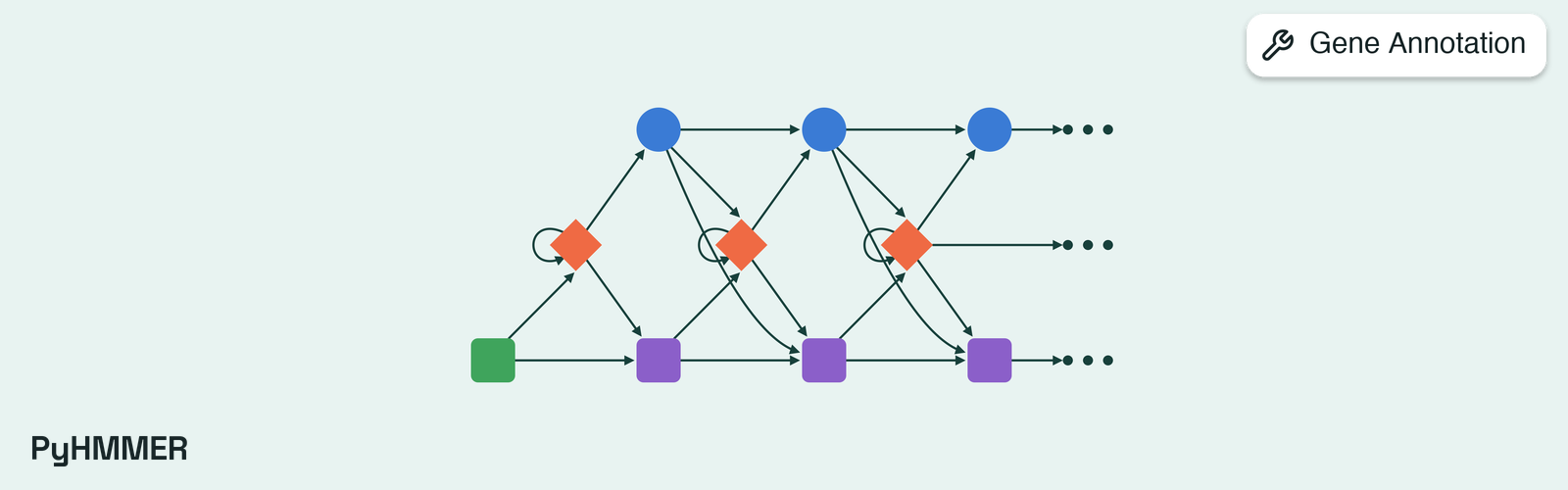
PyHMMER (Larralde & Zeller, 2023) is a Cython binding to the HMMER C API that ships the HMMER source itself, so a singlepip install provides both the Python interface and the compiled search engine. The underlying HMMER3 algorithm (Eddy, 2011) builds a profile hidden Markov model from a multiple sequence alignment, where each match state stores position-specific emission probabilities and the transitions between states model insertions and deletions. Search proceeds through a cascade of accelerated filters: a SIMD-vectorised multiple-segment Viterbi (MSV) filter, a vectorised Viterbi filter, and a Forward/Backward filter, each tightening the candidate set before the final scored alignment. Each hit carries a database-size-independent bit score together with an E-value derived from extreme-value-distribution theory. The E-value calibrates the expected number of false-positive hits at that bit score for the database being searched.
Profile HMMs detect homology that pairwise methods such as BLAST miss because they encode an entire family’s position-specific conservation pattern rather than the similarity of two sequences alone. HMMER3 brought profile-HMM search within roughly the runtime envelope of BLAST while keeping that sensitivity advantage. PyHMMER preserves the algorithm exactly and adds Python-native multithreading, in-memory HMM and sequence handles, and structured result objects. Coordinates returned for HMM matches, target alignments, and envelopes are reported as 1-indexed, inclusive intervals to match biological residue selection conventions.
Learning Resources
- pyhmmer documentation (Martin Larralde) - the canonical API reference, with worked examples for every binding and a guide to feeding HMM and sequence files in and out of memory.
- HMMER User’s Guide (The Eddy/Rivas Laboratory, Harvard) - reference for the HMMER 3 command-line surface, the MSV/Viterbi/Forward filter cascade, E-value statistics, and the gathering/noise/trusted cutoff system used by Pfam HMMs.
- Pfam (via InterPro) (EMBL-EBI) - the standard curated HMM library that ships gathering, noise, and trusted cutoffs, and the typical target database for
hmmscandomain annotation.
Tools
PyHMMER Profile Search (pyhmmer-hmmsearch)
Searches one or more HMM profiles against a set of protein sequences and returns the sequences (and the per-domain alignments within them) that match each profile.API Reference
Source
Input: PyHmmsearchInput
Input: PyHmmsearchInput
string | string
required
Path to an HMM file containing one or more profile HMMs. The file should be in HMMER3 format (typically
.hmm extension). Can contain multiple HMM profiles; all will be searched against the target sequences.List[string]
required
Target protein sequences to search. Inherited from
PyHmmerInput. Can be a single sequence string or a list of sequence strings. Source
Config: PyHmmsearchConfig
Config: PyHmmsearchConfig
string
Use the HMM’s stored bit-score cutoff in place of E-value reporting.
gathering is the Pfam-curated default for inclusion; noise is the most permissive; trusted is the strictest. None = use E-value/score thresholds. Default: None. Pyhmmer raises MissingCutoffs if the HMM file lacks the requested cutoff line — set None for HMMs without curated thresholds.integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cpu"
Device to run the tool on.
integer
default:"3600"
Maximum execution time in seconds.
None waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.integer
default:"0"
CPU threads (0 = auto). Inherited from
PyHmmerConfig.number
default:"10.0"
Sequence-level E-value cap to report. Inherited from
PyHmmerConfig.number
Sequence-level bit-score floor. Inherited from
PyHmmerConfig.number
default:"10.0"
Per-domain E-value cap to report. Inherited from
PyHmmerConfig.number
Per-domain bit-score floor. Inherited from
PyHmmerConfig.number
default:"0.01"
Sequence-level E-value cap for inclusion. Inherited from
PyHmmerConfig.number
default:"0.01"
Per-domain E-value cap for inclusion. Inherited from
PyHmmerConfig.number
Effective database size. Inherited from
PyHmmerConfig.number
Significant hit count. Inherited from
PyHmmerConfig.boolean
default:"False"
Disable MSV/Vit/Fwd filters. Inherited from
PyHmmerConfig.Applications
Use this when the question is “which proteins belong to family X.” Build or download an HMM for a family of interest, then sweep a proteome, a metagenome, or a designed library to enumerate members and pull out their domain coordinates for downstream filtering or alignment.Usage Tips
bit_cutoffs="gathering"activates the Pfam-curated thresholds and replaces the E-value filter. Each Pfam HMM ships a hand-curated gathering (--cut_ga) cutoff that defines family membership, together with the auto-derived noise (--cut_nc) and trusted (--cut_tc) cutoffs that bracket the curated set. Use"gathering"for routine Pfam annotation; ad-hoc HMMs without stored cutoffs raiseMissingCutoffs.- The default
evalue_threshold=10.0is intentionally permissive. Tighten to0.001for confident annotation or1e-10for stringent homology detection; loose thresholds are useful only when you plan to post-filter onincludedordomain_included.
PyHMMER HMM Scan (pyhmmer-hmmscan)
Searches one or more query protein sequences against an HMM database and returns the profiles that match each query.API Reference
Source
Input: PyHmmscanInput
Input: PyHmmscanInput
string | string
required
Path to an HMM database file containing multiple profile HMMs. The file should be in HMMER3 format and typically represents a comprehensive database like Pfam. All HMMs in the database will be searched against the query sequences.
List[string]
required
Query protein sequences to search. Inherited from
PyHmmerInput. Can be a single sequence string or a list of sequence strings. Source
Config: PyHmmscanConfig
Config: PyHmmscanConfig
string
Use the HMM’s stored bit-score cutoff in place of E-value reporting.
gathering is the Pfam-curated default for inclusion; noise is the most permissive; trusted is the strictest. None = use E-value/score thresholds. Default: None. Pyhmmer raises MissingCutoffs if the HMM file lacks the requested cutoff line — set None for HMMs without curated thresholds.integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cpu"
Device to run the tool on.
integer
default:"3600"
Maximum execution time in seconds.
None waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.integer
default:"0"
CPU threads (0 = auto). Inherited from
PyHmmerConfig.number
default:"10.0"
Sequence-level E-value cap to report. Inherited from
PyHmmerConfig.number
Sequence-level bit-score floor. Inherited from
PyHmmerConfig.number
default:"10.0"
Per-domain E-value cap to report. Inherited from
PyHmmerConfig.number
Per-domain bit-score floor. Inherited from
PyHmmerConfig.number
default:"0.01"
Sequence-level E-value cap for inclusion. Inherited from
PyHmmerConfig.number
default:"0.01"
Per-domain E-value cap for inclusion. Inherited from
PyHmmerConfig.number
Effective database size. Inherited from
PyHmmerConfig.number
Significant hit count. Inherited from
PyHmmerConfig.boolean
default:"False"
Disable MSV/Vit/Fwd filters. Inherited from
PyHmmerConfig.Applications
Use this when the question is “what does this protein contain.” Run a query proteome against Pfam to annotate each protein with its domain architecture, then filter ondomain_included to keep curated hits.Usage Tips
- Pick
hmmscanversushmmsearchby what you are querying with.hmmscantakes sequences as queries and a database of HMMs as the target;hmmsearchis the reverse. For one or a few queries against Pfam,hmmscanis the natural choice; for one HMM against a large sequence database,hmmsearchis much faster. bit_cutoffs="gathering"applies here too and is the recommended Pfam annotation default. As withhmmsearch, the cutoff is read from the HMM file and ad-hoc HMMs without stored cutoffs will fail withMissingCutoffs.
PyHMMER Single-Sequence Protein Search (pyhmmer-phmmer)
Searches one or more protein query sequences against a target protein database by building a temporary HMM around each query.API Reference
Source
Input: PyPhmmerInput
Input: PyPhmmerInput
List[string]
required
Target protein sequences to search against. Can be a single sequence string or a list of sequence strings. The query sequences will be compared against these targets.
List[string]
required
Query protein sequences. Inherited from
PyHmmerInput. Can be a single sequence string or a list of sequence strings. These sequences will be used to build temporary HMM profiles on-the-fly. Source
Config: PyHmmerConfig
Config: PyHmmerConfig
integer
default:"0"
CPU threads; 0 = auto-detect. Default 0.
number
default:"10.0"
Sequence-level E-value cap to report. Default 10.0.
number
Sequence-level bit-score floor. Overrides E-value when set. Default None.
number
default:"10.0"
Per-domain E-value cap to report. Default 10.0.
number
Per-domain bit-score floor. Overrides domain E-value when set. Default None.
number
default:"0.01"
Sequence-level inclusion E-value. Default 0.01.
number
default:"0.01"
Per-domain inclusion E-value. Default 0.01.
number
Effective database size for E-value calc. None = use the actual target count.
number
Significant hit count for domain E-value calc. None = use actual.
boolean
default:"False"
Disable MSV/Vit/Fwd heuristic filters. Slower but maximally sensitive. Default False.
integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cpu"
Device to run the tool on.
integer
default:"3600"
Maximum execution time in seconds.
None waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.Applications
Use this for HMM-grade sensitivity when no pre-built profile is available. Typical workflows include finding remote homologs of a newly characterised protein in a reference proteome and running a sequence-based homology pass when the family of interest is too narrow or too new to have a curated HMM.Usage Tips
- A single-query, single-target search will not converge.
phmmerbuilds the HMM from the query against the target database’s residue statistics; a database of one sequence has no background to estimate against. Usephmmerwith a real target proteome, not a synthetic pair.
PyHMMER Nucleotide Search (pyhmmer-nhmmer)
Searches nucleotide query sequences against a nucleotide target database with the same profile-HMM machinery used for proteins.API Reference
Source
Config: PyNhmmerConfig
Config: PyNhmmerConfig
enum
default:"both"
Strand to search.
both (default) runs the forward strand and its reverse complement; watson runs only the forward strand; crick runs only the reverse complement.Available options: both, watson, crickinteger
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cpu"
Device to run the tool on.
integer
default:"3600"
Maximum execution time in seconds.
None waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.integer
default:"0"
CPU threads (0 = auto). Inherited from
PyHmmerConfig.number
default:"10.0"
Sequence-level E-value cap to report. Inherited from
PyHmmerConfig.number
Sequence-level bit-score floor. Inherited from
PyHmmerConfig.number
default:"10.0"
Per-domain E-value cap to report. Inherited from
PyHmmerConfig.number
Per-domain bit-score floor. Inherited from
PyHmmerConfig.number
default:"0.01"
Sequence-level E-value cap for inclusion. Inherited from
PyHmmerConfig.number
default:"0.01"
Per-domain E-value cap for inclusion. Inherited from
PyHmmerConfig.number
Effective database size. Inherited from
PyHmmerConfig.number
Significant hit count. Inherited from
PyHmmerConfig.boolean
default:"False"
Disable MSV/Vit/Fwd filters. Inherited from
PyHmmerConfig.Applications
Use this to find homologs of transposable elements, non-coding RNAs, regulatory elements, and other nucleotide features that diverge fast enough to slip past direct sequence alignment. Pair it with Dfam - the curated profile-HMM library of transposable-element families that was co-designed with nhmmer - or with custom-built nucleotide HMMs when annotating genomes and metagenomic contigs.Usage Tips
stranddefaults to"both"and searches the forward and reverse-complement strands. Set"watson"to restrict to forward or"crick"to restrict to reverse-complement when the orientation of a hit is meaningful (e.g., on annotated coding strands).
PyHMMER Iterative Protein Search (pyhmmer-jackhmmer)
Performs iterative protein-sequence search against a target protein database, rebuilding the HMM from each round’s included hits to extend the search outward across remote homologs.API Reference
Source
Config: PyJackhmmerConfig
Config: PyJackhmmerConfig
integer
default:"5"
Maximum jackhmmer iterations; stops early on convergence. Default 5.
integer
default:"0"
Verbosity level (0=quiet, 1=info, 2=debug, 3=raw subprocess stderr).
True is coerced to 1 and False to 0.string
default:"cpu"
Device to run the tool on.
integer
default:"3600"
Maximum execution time in seconds.
None waits indefinitely.integer
Random seed. When set, tools run reproducibly up to small GPU float noise (see
BaseToolOutput.approx_equal), and the seed participates in cache keys. When None, cacheable seed-sensitive tools skip cache until seeded.integer
default:"0"
CPU threads (0 = auto). Inherited from
PyHmmerConfig.number
default:"10.0"
Sequence-level E-value cap to report. Inherited from
PyHmmerConfig.number
Sequence-level bit-score floor. Inherited from
PyHmmerConfig.number
default:"10.0"
Per-domain E-value cap to report. Inherited from
PyHmmerConfig.number
Per-domain bit-score floor. Inherited from
PyHmmerConfig.number
default:"0.01"
Sequence-level inclusion E-value. Inherited from
PyHmmerConfig. Critical for jackhmmer — the included set seeds the next iteration’s HMM.number
default:"0.01"
Per-domain inclusion E-value. Inherited from
PyHmmerConfig.number
Effective database size for E-value calc. Inherited from
PyHmmerConfig.number
Significant hit count for domain E-value. Inherited from
PyHmmerConfig.boolean
default:"False"
Disable MSV/Vit/Fwd filters. Inherited from
PyHmmerConfig.Applications
Use this when you need to reach divergent family members that a single-passphmmer would miss, for example when seeding a new family from one characterised representative or expanding a manually curated set to its full evolutionary breadth.Usage Tips
inclusion_evalue_thresholdis the lever that controls iterative drift. Each iteration rebuilds the HMM from hits that pass the inclusion thresholds (--incE/--incdomE, defaults0.01). A looser inclusion threshold pulls in more sequences per round and increases the risk of pulling in unrelated families; tighten it when iterations start drifting.max_iterationsdefaults to 5 and the search exits early on convergence. Raising it rarely helps if the search has already converged on a stable set, and a higher cap multiplies runtime on long-running jobs.
Toolkit Notes
These apply to every PyHMMER tool in this toolkit (pyhmmer-hmmsearch, pyhmmer-hmmscan, pyhmmer-phmmer, pyhmmer-nhmmer, pyhmmer-jackhmmer).
- Runs on CPU with SIMD acceleration. The HMMER3 filter cascade is SIMD-vectorised on x86 platforms. pyhmmer compiles HMMER from source at install time and inherits whatever instruction sets the build host exposes, with no GPU acceleration to enable.
- Self-contained after install. The HMMER C library is compiled into the PyHMMER wheel, so no separate HMMER install or PATH lookup is needed; HMM databases such as Pfam-A still have to be downloaded separately.
num_threadsparallelises within a single search. Default0auto-detects the available cores. Memory scales with HMM database size; Pfam-A (around 20,000 HMMs) needs roughly 2 GB of RAM held resident.- Reporting versus inclusion thresholds are independent filters.
evalue_threshold/score_threshold(and theirdomain_*siblings) control what appears in the output, whileinclusion_evalue_thresholdmarks the stricter “trusted” subset via theincludedanddomain_includedflags.jackhmmerseeds the next iteration’s HMM from the included set, so the inclusion threshold drives iterative behaviour while reporting only affects what is returned.
Infrastructure Guides
The following guides cover how to run tools efficiently and at scale.Tool Persistence
Keep a tool’s model warm across calls instead of reloading it every invocation.
Device Management
How GPUs are allocated to tools and how to target specific devices.
Parallel Execution
Fan a batch of inputs out across multiple GPUs.